 |
COMPUTATIONAL BASICS UNDERLYING SYNTHETIC STRATEGIES |
| << SOME CONCEPTUAL DEVELOPMENTS IN SYNTHESIS IN CHEMISTRY |
Chapter
- 17
COMPUTATIONAL
BASICS UNDERLYING SYNTHETIC
STRATEGIES
S.
Sabiah
KEY
WORDS: Computation,
Synthetic strategy, Synthesis
design, Computer-
assisted
synthesis
INTRODUCTION:
Synthetic
strategy is an important stepping stone
for scientific community. It
has been
found
so crucial and innovative in
the four mainstream of
research namely,
1.
Observational Science
2.
Experimental Science
3.
Theoretical Science
4.
Computational Science
Since
the topic of choice is on
computational, I would like to
draw few lines about
the
computational
science. Computational
science (or
scientific
computing)
is the field
of
study concerned with constructing
mathematical models and
numerical solution
techniques
and using computers to analyze
and solve scientific, social
scientific and
engineering
problems. In practical use, it is
typically the application of
computer
simulation
and other forms of
computation to problems in various
scientific
disciplines.
It uses everything that scientists
already know about a problem
and
incorporates
it into a mathematical problem
which can be solved. The
mathematical
model
which then develops gives
scientists more information about
the problem.
Computational
Science is beneficial for two
main reasons:
1.
It is a cheaper method of conducting
experiments.
2.
It provides scientists with extra
information which helps them
to better
plan
and hypothesizes about
experiments.
Due
to these reasons, computation
has gained much attention in
almost all fields of
science
which are listed below under
computational disciplines.
COMPUTATIONAL
DISCIPLINES
Bioinformatics
·
Cheminformatics
·
Chemometrics
·
Computational
biology
·
17.2
Computational
Basics Underlying Synthetic
Strategies
Computational
chemistry
·
Computational
economics
·
Computational
electromagnetics
·
Computational
engineering
·
Computational
fluid dynamics
·
Computational
mathematics
·
Computational
mechanics
·
Computational
physics
·
Computational
statistics
·
Environmental
simulation
·
Financial
modeling
·
Geographic
information system
(GIS)
·
High
performance computing
·
Machine
learning
·
Network
analysis
·
Numerical
weather prediction
·
Pattern
recognition
·
Since
the main focus of the
chapter is on synthetic
strategies in chemistry,
it is
likely
to be elaborated towards chemical
aspects. In the field of
chemistry, we all
know
that the beginning of the
organic synthesis is started with the
preparation of urea
by
Wohler in 1828. It is only in
1967 that a systematic analysis of
synthesis towards
the
direction of computer-assisted synthesis design
(CASD) was reported by E.
J.
Corey
[1]. Since then the
computational methods in chemistry has
been growing in
many
dimensions and every
scientific paper pays attention to
computational support
in
addition to the experimental
evidences. It has also become a useful
way to
investigate
materials that are too
difficult to find or too
expensive to purchase. It
helps
chemists
to make predictions before
running the actual
experiments so that they
can
be
better prepared for making
observations. Hence, we can say
that computational
chemistry
is partly assisting the basic chemical
problems deal with synthesis,
structure
and
spectroscopy of materials and indeed
important to discuss
further.
Computational
Chemistry
Computational
chemistry is a branch of chemistry
that generates data,
which
complements
experimental data on the structures,
properties and reactions
of
substances.
It can in some cases predict
hitherto unobserved chemical
phenomena. It
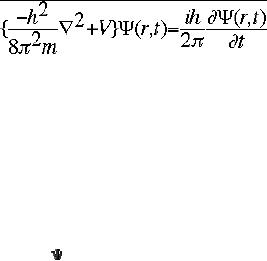
Synthetic
Strategies in Chemistry
17.3
is
widely used in the design of
new drugs and materials.
The calculations are
based
primarily
on Schrödinger's equation (eqn.
1).
.................eqn
1
The
symbol ψ is a mathematical function
that calculates the strength of
the deBroglie
wave
at various positions in space.
The rest of the components
are as follows:
h
=
Planck's constant
m
=
the mass of the
particle
Ć
= a partial differential operator
called the Laplacian
operator
V=the
potential energy
=psi,
the wave function
i
=the
square root of -1
The
final form of equation 1
is Hψ =
Eψ
Where
H = Hamiltonian Operator; E = total
energy of the system
Computational
chemistry is particularly useful
for determining
molecular
·
properties
which are inaccessible
experimentally and for
interpreting
experimental
data
With
computational chemistry, one
can calculate:
·
electronic
structure determinations
o
geometry
optimizations
o
frequency
calculations
o
transition
structures
o
protein
calculations, i.e.
docking
o
electron
and charge distributions
o
potential
energy surfaces (PES)
o
rate
constants for chemical
reactions (kinetics)
o
thermodynamic
calculations- heat of reactions, energy
of activation
o
There
are three main types of
calculations:
·
1.
Ab Initio: (Latin for "from
scratch") a group of methods in
which
molecular
structures can be calculated using
nothing but the
Schroedinger
equation, the values of the
fundamental constants and
the
atomic
numbers of the atoms present
17.4
Computational
Basics Underlying Synthetic
Strategies
2.
Semi-empirical: techniques use
approximations from
empirical
(experimental)
data to provide the input
into the mathematical
models
3.
Molecular mechanics: uses classical
physics to explain and
interpret
the
behavior of atoms and
molecules
Currently,
there are two ways to
approach chemistry problems: computational
quantum
chemistry and
non-computational
quantum chemistry. Computational
quantum
chemistry is primarily concerned with
the numerical computation
of
molecular
electronic structures by ab
initio and
semi-empirical techniques and
non-
computational
quantum chemistry deals with
the formulation of analytical
expressions
for
the properties of molecules
and their reactions.
Examples of such properties
are
structure
(i.e. the expected positions
of the constituent atoms), absolute
and relative
(interaction)
energies, electronic charge distributions,
dipoles and higher
multipole
moments,
vibrational frequencies, reactivity or
other spectroscopic quantities,
and
cross
sections for collision with
other particles.
The
methods employed cover both
static and dynamic
situations. In all cases
the
computer
time increases rapidly with
the size of the system
being studied. That
system
can be a single molecule, a
group of molecules or a solid.
The methods are
thus
based on theories which
range from highly accurate,
but are suitable only
for
small
systems, to very approximate,
but suitable for very
large systems. The
accurate
methods
used are called ab initio
methods, as they are based
entirely on theory
from
first
principles. The less
accurate methods are called
empirical or semi-empirical
because
some experimental results, often
from atoms or related
molecules, are used
along
with theory.
There
are two different aspects to
computational chemistry:
Computational
studies can be carried out in
order to find a starting
point for a
·
laboratory
synthesis, or to assist in understanding
experimental data, such as
the
position and source of spectroscopic
peaks.
Computational
studies can be used to predict
the possibility of so far
entirely
·
unknown
molecules or to explore reaction
mechanisms that are not
readily
studied
by experimental means.
To
perform these computational
simulations or calculations, super
computers with
high
performance computing facility is
needed.
Synthetic
Strategies in Chemistry
17.5
Impact
of High Performance Computing
The
amount of chemical information is
quite large and calls
for computer as the
main
source
for storage. With
presently
17
million known
compounds
·
500000
new compounds each
year
·
600
000 chemistry-related publications
annually
·
An
overview and access to all
this information can only be
maintained by electronic
means.
Thus,
databases on chemical information
play a major role in present
day research and
development.
Databases provide access
to
literature
on 17 million compounds
·
factual
data on 7 million organic
compounds
·
several
million reactions
·
140000
experimental 3D structures
·
250
000 spectra
·
SOFTWARES
The
use of computers as tools in chemistry
dates back to late 1950s.
The adoption of
FORTRAN
as a scientific programming
language
is
the beginning of
these
studies[2].
Only from around 1980
1990, the user had
access to a variety of
network
based
resources from a single
point of use. A more
sophisticated example is the
use of
Java
to display digital spectral information
derived from an NMR spectrometer,
[3]
and
Java is a computer language developed by
Sun Microsystems for
writing
programs
to run on the web within a
browser. It is used to link
regions of the spectrum
to
specific atoms or residues in a 3D
molecular object. The links
can be bi-directional,
i.e.
clicking on a specified atom will
highlight the spectral
region containing peaks
associated
with that atom.
Chemists
have been some of the most
active and innovative
participants in this
rapid
expansion
of computational science. Some
common computer software
used for
computational
chemistry includes:
Gaussian
03
·
GAMESS
·
MOPAC
·
Spartan
·
17.6
Computational
Basics Underlying Synthetic
Strategies
Sybyl
·
Now
let us move towards the
use of these computational
basics for synthetic
strategy
especially
in basic organic chemistry. Once we
know how it works, it can
be
extendable
to other materials.
1.
IN ORGANIC SYNTHESIS
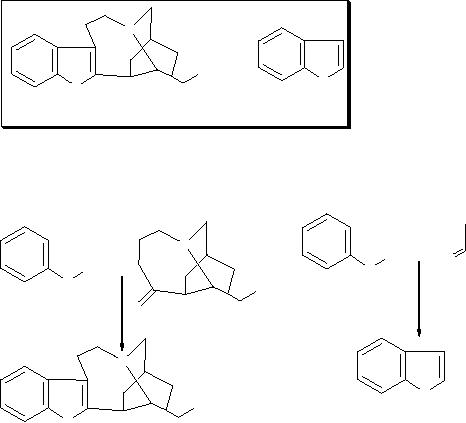
When
a chemist wants to synthesize a compound
of interest, it is called target
molecule
and he
looks in to the target to
identify any known fragment
called
substructure
is
present whose synthesis is already available.
For example, in the
synthesis
of the Ibogamine, chemist perceives
the presence of an indole
nucleus,
whose
synthesis is known (Chemical structures are
shown inside the box).
So, he may
plan
the synthesis similar to Indole as
outlined in Scheme 1.
N
N
N
H
H
Indole
Ibogamine
Scheme
1
Synthesis
of Ibogamine
Synthesis
of Indole
N
NH2 +
O
+
N
NH2
H
N
H
O
N
N
H
N
Indole
H
Ibogamine
A
known starting
material is
very similar to the target
and the problem is 'reduced'
to
find
the reactions which will
convert it to the target.
Despite the similarity of
the
starting
material and the target,
the number of steps to
obtain the target may be
high in
certain
cases which triggered the
venture of using a computer to
solve synthesis
problems
[4]. The number of reactions
being very high (several
thousands), the
computer
should be able to store all
these reactions. It would
never forget them and
it
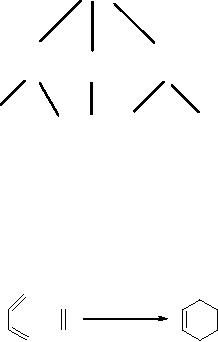
Synthetic
Strategies in Chemistry
17.7
could
generate all the possibilities.
For this purpose, Corey
proposed a general
approach
which is discussed.
Corey's
Approach
Starting
from the target, the
program finds its precursors,
then
each precursor
becomes
a target and new precursors
are generated; the process
is then repeated until
commercial
or simple products are
generated. This approach is
called retro-synthetic
or
antithetic, since it is the reverse of
the synthesis as practiced at the
bench. This
approach
may be visualized by a 'synthesis
tree' (Scheme
2). This approach
should,
formally,
be 'easily' programmed: it necessitates
'only' writing a program
able to
generate
the precursors of a target and
this program is repeated again
and again.
Numerous
programs have been written
after this initial report
and several reviews
on
this
subject have been published
[5].
Scheme
2
Target
First
level
P3
P1
of
precursors
P2
Second
level
P4
P5
P6
P8
P7
of
precursors
The
above tree
points
outline the essential problems
like how to describe a
molecule
and
how to describe a reaction, that
is, how to generate a precursor.
Let us consider
the
classical Diels-Alder reaction,
Scheme
3
dienophile
diene
cyclohexene
2
1
3
The
above equation corresponds to writing of
the reaction in the
synthetic (or
forward)
direction: reacting 1
with
2,
under
appropriate conditions, generates
3.
In the
approach
proposed by Corey, the
program works backward, from
the target to the
precursor:
in the retro-synthetic or antithetic
direction. So the program
has to search
the
substructure
3
in
the target and, if it is present, it
replaces it by the precursor(s)
17.8
Computational
Basics Underlying Synthetic
Strategies
substructure(s),
here 1 + 2. To describe the reaction in
the retro-synthetic
direction,
the
term of 'transform'
has been
proposed [6] and the
use of a double lined
arrow
indicates
this operation (Scheme
4):
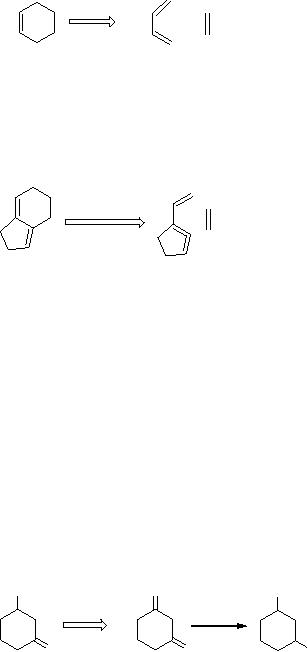
Scheme
4
cyclohexene
dienophile
diene
This
description may look simple
for a computer, but fails
for certain circumstances.
For
example,
Scheme
5
5
4
If
4 is the target then the
precursor would be 5, which is an
impossible solution due
to
the
allenic function in a five-membered
ring. But, computer predicts
scheme 5 as a
retro-synthetic
approach which is not
applicable to laboratory synthesis.
Similarly, if
the
computer is taught the
reduction of a keto group
(7) to form an alcohol
(6),
described
in scheme 6, the backward
mode by the transform of
scheme it will
generate
7 as a possible precursor for 6.
This is again a wrong
solution, indeed there
is
another
keto group which will
generally be reduced, so that, in the
laboratory the
result
would not be 6 but 8 (Scheme
6).
Scheme
6
OH
O
OH
O
O
OH
7
6
8
These
two examples clearly
indicate that the computer
should know to discard
wrong
solutions
for a synthetic strategy.
Hence there should be a way
to settle
communication
between the computer and
the chemist for the
following aspects:
I.
Description
of molecules
II.
Description
of reactions
Synthetic
Strategies in Chemistry
17.9
III.
Pruning
the synthesis tree
These
aspects are discussed in the
upcoming section.
I.
Description of Molecules
(a)
Use of Connectivity
Table
The
molecules of interest are
first described by connectivity
tables which
list in
several
arrays the atoms and the
bonds of the target. An
example is given with
atom
labeling
(bold) and nature of bonds
(normal numbers) for
compound A.
2
O
1
1C
3
5
4
3
2
C
N
H3C
4
compund
A
Then
the connectivity is given by
atomic table and bond
table as described in
Tables
17.1
and 17.2. These tables are
not given in this form to
the computer. Actually
the
chemist
draws the structure on the
screen using a mouse [7],
and a program takes
care
of
transforming this graphic
structure into a connectivity
table.
Table
17.1. Atomic table for
compound A
Bonds
Number
of hydrogen
Atom
number
Atom
type
Neighbors
number
number
atoms
1
C
2
3
4
1
2
3
0
2
O
1
1
0
3
C
1
2
3
4
C
1
5
3
4
0
5
N
4
4
0
This
description is a topological
one,
but when a chemist looks at
a target he does not
only
see a succession of atoms and
bonds. He automatically perceives
structural
features,
such as functional groups,
rings, stereo-centers, and their
relative positions.
The
information is central to finding
the solutions of the
problem. The computer
also
needs
to be taught this chemical
perception of the target. So,
when the drawing of
the
target
is done and the connectivity
tables have been established,
before starting the
retro-synthetic
process, the program
searches for these characteristics
and stores them
17.10
Computational
Basics Underlying Synthetic
Strategies
in
a binary array which is a
kind of identity card of the
target. An example of a
partial
binary
array for structure A is
given in Table 17.3.
Table
17.2. Bond table for
compound A
Bond
number
Atom
1
Atom
2
Bond
type
1
1
2
2
2
1
3
1
3
1
4
1
4
4
5
3
Table
17.3. Binary array for
compound A
Groups
C
N
O
CH
CH2
CH3
Ketone
Nitrile
Cyclic
atom
atom
number
1
1
0
0
0
0
0
1
0
0
2
0
0
1
0
0
0
0
0
0
3
1
0
0
0
0
1
0
0
0
4
1
0
0
0
0
0
0
1
0
5
0
1
0
0
0
0
0
0
0
With
the topological tables and
this analytical one, the
program gains a
chemical
perception
of the target.
(b)
Use of Matrices
In
1971 Ugi and Gillespie [8]
proposed the concept of 'BE'
(for bond-electron)
matrices
to describe molecules. 'BE' matrices were
then transformed into
connection
tables.
In this model the element
(i, j)
of
the matrix corresponds to the
bond order
between
atoms i and j
(l =
single bond, 2 = double
bond, ...) and the
diagonal elements
(i,
i) describe the free valence
electrons of atom i. Compound A is
described by the
matrix
of equation (1):
Synthetic
Strategies in Chemistry
17.11
1
2
3
4
5
1
0
2
1
1
0
2
2
2
0
0
0
3
1
0
0
0
0
(1)
4
1
0
0
0
3
5
0
0
0
3
1
(c)
Use of Numerical Linear
Notation
In
1971, Hendrickson proposed a
logical description of structures and
reactions by a
simple
mathematical model [9] which
has been developed and used
in the program
SYNGEN.
This mainly focuses on
carbon skeleton as shown for
a three carbon
system
of compound B. In the original
system four kinds of attachments to
any carbon
are
described and counted: H for
attachment of hydrogen, or
electropositive atoms; R
for
σ-bond to another carbon; Π
for -bond to another carbon
and Z for a bond (σ
or
)
to an electronegative heteroatom (N, O,
S, X). The H, R, Π and Z are
notations
used
in the program. How many
numbers of such attachments is described by h,
σ,
,
z,
respectively. The functionality
(f)
at a carbon site is defined as
the sum of z and ,
and
the character (c)
of a
carbon site is c = l0 σ + f.
In
SYNGEN the functional groups
on each carbon atom are
abstracted with the
two
digits
z and ; for atom 2 of
Scheme 7, the z list is 11,
for atom 3 it is 30.
1
Cl
H2C
C
Scheme
7
C
N
2
3
Compound
B
c
z
σ
atom
f
1
11
1
0
1
1
2
22
1
1
2
2
3
13
0
3
1
3
II.
Coding of a Particular
Reaction
The
three main systems described
above lead to three main
methods for coding
reactions
in CASD programs. The
description of reactions leads also to
two families
of
CASD programs: empirical and
non-empirical. The empirical
programs are based
17.12
Computational
Basics Underlying Synthetic
Strategies
on
known reaction libraries.
The advantage of such an
approach is that the
programs
predict
syntheses which have great
chances of feasibility provided
that specific
structural
features do not strongly interfere
with one of the proposed
reactions. On the
other
hand, these programs cannot
suggest totally new synthesis
reactions. Further,
the
number of reactions to code is very
large; theoretically, all
known reactions
should
be
coded in the reaction
files.
In
the non-empirical programs
reactions are coded in a
logical, mathematical or
general
way, in order to describe the
maximum of reactions with a
minimum of
principles.
The aim is also to have a
system able to propose new
syntheses, even new
reactions
if they have not yet been
described in the
literature
(a)
The Transform
Approach
As
indicated above, the word
'transform' is employed to describe a
reaction in the
retro-synthetic
direction. The aim of a CASD
program is to generate the precursors
of
a
target. To do this, a program
has three main tasks to
perform:
-
Search for a substructure
which describes the
transform (called synthon or
retron)
-
Generation of the precursors
-
Evaluation of the validity of
the solutions.
The
main differences between the
various programs come from
the evaluation step,
which
may be more or less accurate. In
SOS program, a full graphic
interface has
been
developed to input transform
and evaluation tests. For
example, the user
draws
the
retron and its precursor
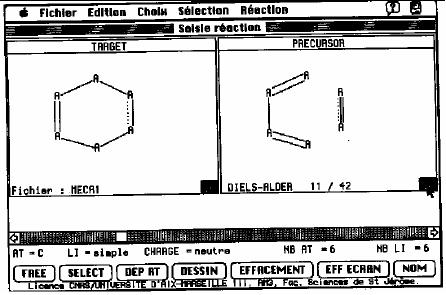
with the mouse (Fig.
17.1). Let us consider the
example of
the
Diels-Alder reaction. Symbol A
stands for any atom in
order to generalize
the
reaction.
Since the diene may react
with a double or a triple
bond, one may use
the
simple/double
bond in the target and
double/triple bond in the
precursor (bonds with
dotted
lines).
Synthetic
Strategies in Chemistry
17.13
Fig.
17.1. Representation of an active
window from SOS
program
To
enter a test, the chemist
selects this option in the
reaction menu and indicates
that
the
test must be applied on the
target. The atoms and
the bonds are numbered
and the
user
may enter the test by
clicking on the buttons at
the bottom of the screen.
The test
allows
for a solution such as that
if atom 4 is sp2 and bond 3 is a fused
bond then the
reaction
is impossible. This program
also allows graphical tests:
the user draws a
substructure
and the action to perform if
it is present in the scrutinized
target.
In
SOS, coding of reactions by
means of mechanisms (i.e.,
elimination,
nucleophilic
addition, nucleophilic substitution,
etc.) allowed chemists to
generalize
reactions,
to reduce the number of reactions in
the files, and to extend a
scheme to a
new
case even if it has not been
described in the
literature.
(b)
Be-matrices Approach.
Ugi
and Dugundji developed a
mathematical model of constitutional
chemistry [10].
This
model is based on the
concept of isomerism of molecules
which has been
extended
to ensembles of molecules. For
example, a theoretical reaction: A +
B
C
+
D can be seen as the
conversion of an ensemble of molecules (A + B)
into an
isomeric
ensemble (C + D). As an extension, the
discovering of a synthesis:
Target
Precursor
1
Precursor
2
::::
Starting
materials, may be done by
generation of
isomers.
The
description of molecules by means of
matrices allows one to describe
reactions
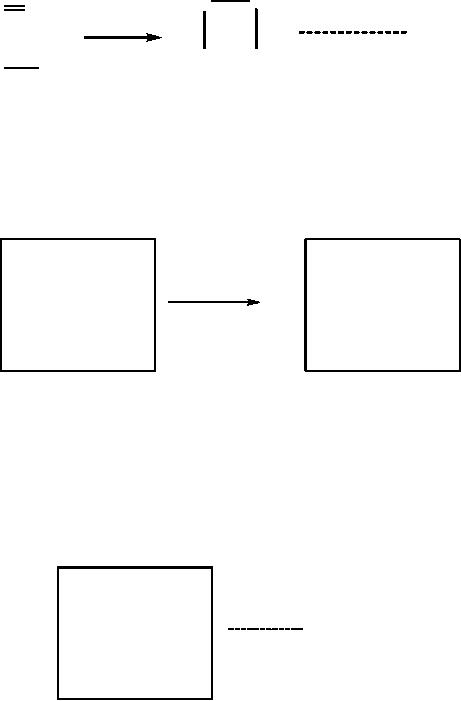
by additions of matrices; let us
take the addition of H-Br on
a double bond
(equation
2).
17.14
Computational
Basics Underlying Synthetic
Strategies
1
2
1
2
H2C
CH2
H2C
CH2
(2)
H
Br
H
Br
4
3
4
3
Let
B be the be-matrix for the
starting materials and E the
be-matrix for the
end
products
(Scheme 8).
Scheme
8
1
2
1
2
3
4
3
4
1
1
0
2
0
0
0
1
0
1
2
0
0
0
2
1
0
1
0
2
0
0
0
1
0
1
0
0
3
3
0
0
1
0
1
0
0
0
4
4
B
E
The
transformation of B into E is defined by
the reaction matrix R of
equation (3)
such
that: B + R = E where off-diagonal
entries Rij
=
Rji =
0, ±l, ±2, ±3, indicate
the
bonds
made or broken. This method
is conceptually attractive because
the retro-
synthesis
is simply: B = E - R.
1
2
3
4
1
0
-1
0
+1
2
-1
0
+1
0
(3)
R=
0
+1
0
-1
3
+1
0
-1
0
4
(c)
The Numerical
Approach
The
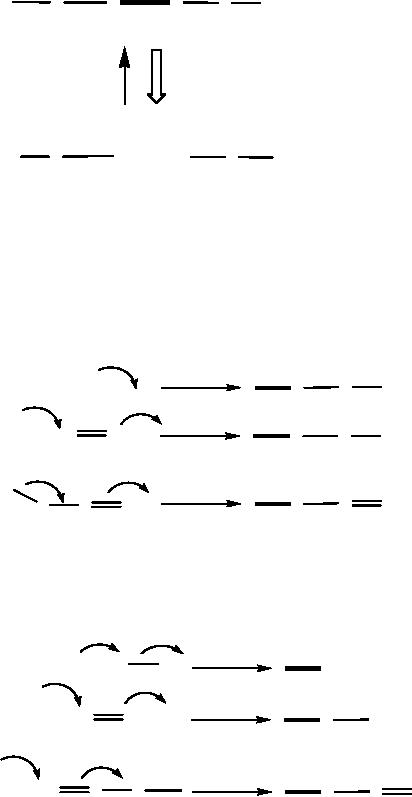
numerical approach is dealt by
different ways by different
programs. The
SYNGEN
program is based upon the
concept of half-reactions; the formation
of a
bond
may be seen as two linked
half-reactions on each side of
the bond (Scheme
9).
Synthetic
Strategies in Chemistry
17.15
Scheme
9
C
C
C
C
C
C
α
γ
α
γ
β
β
C-
+
C
C
C
C
C
α
γ
α
γ
β
β
Electrophilic
Nucleophilic
Half-reaction
Half-reaction
Only
three atoms are considered
around the bond which is
formed, because more
than
three
carbons which change in a half-reaction
are virtually never found
[11]. The
nucleophilic
centers are given in Scheme
10, and the electrophilic
ones in Scheme 11.
Scheme
10
γ
α
β
-
C
C
C
C
Z
C
C
C
Z
C
H
C
C
C
C
C
C
Thus,
combining these three
nucleophilic half-reactions with
the three
electrophilic
half-reactions
produces nine possible full
construction reactions. A reaction
may also
be
described by two letters, the
first for the bond made,
second for that
broken.
Scheme
11
γ
α
β
C
Z
C
H
C
C
CH
C
C
C
C
Z
C
C
C
For
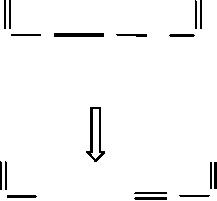
the Michael reaction (Scheme
12), this concept yields
for atom 2: formation of
an
σ
bond (+ R) and loss of
hydrogen (- H), the reaction
on this carbon is designated
by
RH;
for atom 3: formation of an σ
bond and loss of a bond: R
Π and for atom 4: H
Π.
17.16
Computational
Basics Underlying Synthetic
Strategies
Scheme
12
O
O
C
C
C
CH
C
1
2
3
4
5
O
O
+
C
C
C
C
CH
1
2
3
4
5
Since
there are four kinds of
bond (H, R, Π, Z), there are
16 possible unit exchanges
per
carbon. This code allows a
simple classification of reactions,
for example ZH
represents
oxidation reactions (CH
→C-OH), HZ represents reduction,
the opposite of
the
previous one; addition
reactions on a carbon-carbon double
bond are: HΠ,
RΠ,
ZΠ,
etc. This systematic definition of
organic reactions provided a
basis for
developing
COGNOS, a program for
organizing and retrieving
reactions in a large
database
[12].
III.
Pruning the Synthesis
Tree
The
descriptions which are given
above concern the coding of
one reaction and how
a
precursor
may be generated. In a basic
retro-synthesis program, each
reaction of the
file
(or of the files) is applied
in turn for generating precursors
and building, step by
step,
the synthesis tree. As indicated,
the number of solutions
found may be large
and
it
is necessary to develop methods that
are strategies, in order to reduce this
tree and
to
try to select the best
solutions.
The
search of the key-step in a synthesis
is, rather, a fundamental
step. When it
has
been found, one may say
that a general plan of the
synthesis has been found.
This
search
has been done in a very
simple way in the SAS
program: this program
simply
deleted
one or several bonds in the
target, suggesting ideas of synthesis.
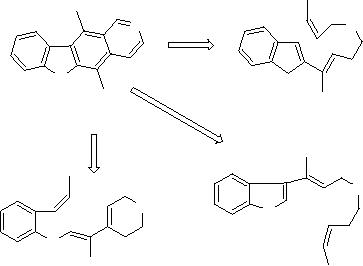
For example,
in
the case of ellipticine
(Compound D), it suggested
that several internal
Diels-Alder
reactions
are involved. Solutions similar to
E
and
F
have been
subsequently and
independently
found experimentally by others groups of
researchers [13]
(Scheme
13).
Synthetic
Strategies in Chemistry
17.17
Scheme
13
N
NH
N
H
E
D
NH
NH
N
H
N
H
F
G
The
other Strategies also
include starting material
oriented strategy,
topological
Strategies,
stereo-chemical Strategies and
Tactical Combinations of
Transforms
Strategy
for pruning the synthesis
tree for organic compounds.
The idea derived
from
this is also applicable to
synthesis of Inorganic
metal complexes,
Energetic
materials, Bio-molecules and drug design
which
are briefly
described
in the following
section.
2.
INORGANIC MATERIALS
(a)
Structure Identification:
In
principle, systems for
representing organic compounds
can be applied to
inorganic
substances,
as far as compounds are concerned in
which the atoms are
connected by
covalent
bonds. The Chemical
Abstracts Service (CAS)
registry system uses
the
notion
of a mixture to accommodate substances
without structure, e.g.,
alloys,
whereas
covalently bonded inorganic
compounds represented by the connection
table
(CT)
based system of CAS. Salts
are represented as mixtures of the
possibly
structured
ions constituting the
salt.
In
organic compounds an atom is
generally connected to at most
four
neighbouring
atoms where as in inorganic
compounds larger coordination numbers
is
found
and, consequently, a more
involved stereochemistry is
encountered.
Furthermore,
one has to deal with
more complicated types of
chemical bonds and
sometimes
there are well defined
substances for which it is
not obvious how to draw
a
structure
at all. Whereas the subjects of
organic chemistry are
exclusively covalently
bonded
compounds of carbon, inorganic
compounds furthermore comprise
ionically
17.18
Computational
Basics Underlying Synthetic
Strategies
bonded
substances like salts, alloys
and glasses, different kinds
of solutions, minerals,
etc.
Therefore a representation of inorganic
compounds must provide some
means to
deal
not only with covalently
bonded compounds but also
with substances which
belong
to those other types [14].Thus,
the generalization leads the
concept of multi-
component
systems in which each
component consists of one or
several fragments.
The
fragments turn may be
structured or not, i.e., it
may be possible to draw
a
structure
diagram for them or not. In
the structure storage system of
the Gmelin
Database,
compounds without a structure are
taken into account by a
tabular
representation,
the inorganic structure tables
(1ST). The hierarchy is
shown in Fig.
17.4.
Substance
Figure
4
Component
1
Component
n
Fragment
n
Fragment
1
Yes
No
Structure
?
CT
IST
(b)
Methodology
Molecular
mechanics (MM) calculations have become
increasingly important in
understanding
the structures and steric
interactions that occur in
inorganic,
bioinorganic,
and organo-metallic compounds.
Since 1984, the growing
use of the
MM
model in inorganic chemistry
has been documented in a number of
reviews [15].
Metal
complexes that have been
treated with MM typically
involve one metal
and
multidentate
organic ligands. The common
functional groups found in
these ligands
include
amines, imines, pyridines, amino acids,
alcohols, ethers, carboxylic
acids,
thiols,
sulfides, and phosphines.
Many MM models, developed
originally for
Synthetic
Strategies in Chemistry
17.19
application
to organic molecules, have
the capability to treat the
ligand part of the
metal
complex. These organic MM
models have been extended to
include metal-
ligand
interactions with the
addition of relatively few terms.
Various approaches
are
distinguished
by the types of metal-dependent
interaction that are
used.
The
two bonded methods in common
use are the valence
force field (VFF)
method
and the points-on-a-sphere (POS)
method. In both methods the
metal ion, M,
is
formally connected to the ligand
donor atoms, L. In a normal organic MM
program,
this
connection will create M - L bonds, M -
L- X and L-M-L bond angles,
M-L-X-X
and
X-M-L-X torsions, and M - X non-bonded
interactions. The user decides
which
of
these interactions will be used in
the calculation through a
choice of parameters.
The
VFF and POS methods
differ only in the treatment
of L- M - L angles. In the
VFF
method, L- M - L bond angle
interactions are used to define a
geometry prefer-
ence
about the metal center. In
the POS method, L- M - L
bond angle interactions
are
not
used and geometry preference
about the metal center
derives primarily from 1,
3
van
der Waals interactions
between the donor atoms. The
third method is
non-bonded
method
where the metal ion is
not formally connected to the
ligand donors; the
metal-
ligand
complex is modeled with a
collection of pair-wise electrostatic
and van der
Waals
interactions. The three methods
which are commonly used
for denoting these
interactions
are summarized in Table
17.4.
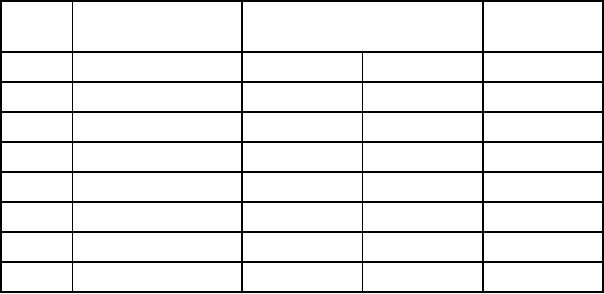
Table
17.4. Methods to extend MM
models for metal
complexes
No
Interaction
Bonded
Non-bonded
VFF
POS
Ionic
1
M-L
stretch
yes
yes
no
2
M-L-X
bend
yes
yes
no
3
L-M-L
bend
yes
no
no
4
L-L
non-bonded
no
yes
yes
5
M-L-X-X
torsion
yes
yes
no
6
L-M-L-X
torsion
no
no
no
7
M-L
non-bonded
no
no
yes
8
M-X
non-bonded
no
no
yes
Apart
from this, the geometry
optimization and frequency
analysis will give an
idea
about
the stability of the
molecules for laboratory synthesis. It is
also possible to get
structure-reactivity
relationship for metal
complexes. The concept of
structure-
17.20
Computational
Basics Underlying Synthetic
Strategies
reactivity
relationship implies that
changes in structure should be
quantitatively
reflected
in some measurable reactivity parameters
associated with the
molecule. For
metal
complexes, the capacity of
the ligand structure to
influence chemical
properties
is
measurable in terms of reactivity parameters such as
stability constants, rates of
ligand
dissociation, and reduction
potentials. The influence of
structure on chemical
reactivity
can often be rationalized in
terms of steric and electronic
components which
module
the synthesis of a particular compound.
Same way, the
structure-reactivity
plays
important role in constructing
energetic materials which is
outlined below.
3.
ENERGETIC MATERIALS
Energetic
materials encompass different
classes of chemical compositions of
fuel and
oxidant
that react rapidly upon
initiation and release large
quantities of force
(through
the
generation of high-velocity product
species) or energy (in the
form of heat and
light).
These particular features have been
advantageously employed in a wide
variety
of
industrial and military
applications, but often
these utilizations have not
been fully
optimized,
mainly due to the inability
to identify and understand
the individual
fundamental
chemical and physical steps
that control the conversion
of the material to
its
final products. The
conversion of the material is
usually not the result of a
single-
step
reaction, or even a set of a
few simple consecutive
chemical reactions. Rather,
it
is
an extremely complex process in
which numerous chemical and
physical events
occur
in a concerted and synergistic
fashion, and whose reaction
mechanisms are
strongly
dependent on a wide variety of
factors.
Also,
these processes often occur
under extreme conditions of
temperature and
pressures,
making experimental measurement
difficult. These are but a
few of the
complexities
associated with studies of reactions of
energetic materials that
make
resolving
the individual details so
difficult. These difficulties
have required the
development
of a variety of innovative theoretical
methods, models and
experiments
designed
to probe details of the
various phenomena associated
with the conversion
of
energetic
materials to products
[16].
The
high time and pecuniary
costs associated with the
synthesis or formulation,
testing
and fielding of a new
energetic material has
called for the inclusion
of
modeling
and simulation into the
energetic materials design
process. This has
resulted
in
growing demands for accurate
models to predict properties
and behavior of
notional
energetic materials before
committing resources for
their development.
For
example,
in earlier times, extensive
testing and modification of
proposed candidate
Synthetic
Strategies in Chemistry
17.21
materials
for military applications
could take decades before
the material was
actually
fielded,
in order to assure the
quality and consistent performance of
the Predictive
models
that will allow for
the screening and
elimination of poor candidates before
the
expenditure
of time and resources on synthesis
and testing of advanced
materials
promise
significant economic benefit in
the development of a new
material.
4.
BIO-MOLECULES
Computational
bio-modeling refers
to a type of artificial life
research concerned
with
building computer simulations of
biological systems (bio-modeling).
The
immediate
goal is to understand how
biological entities such as
cells or whole
organisms,
develop, work collectively,
and survive in changing
environments using a
purely
computational model. In order to meet
this challenge we need to establish
the
methodologies
and techniques that will
enable us to gain a system-level
understanding
of
biological processes. These
kinds of models hold great
promise for new
discoveries
in
a wide variety of biological
systems. Once an executable model
has been built of a
particular
system, it can be used to
get a global dynamic picture
of how the system
responds
to various perturbations. In addition,
preliminary studies can be
quickly
performed
using executable models,
saving valuable laboratory
time and resources
for
only
the most promising avenues.
One
way of approaching this
problem is to use ideas and methods
originally
developed
in computer science, mainly in
software and systems
engineering (and in
particular
visual languages and formal
verification) to construct, simulate
and analyze
biological
models. The benefit of
molecular modeling is that it
reduces the
complexity
of
the system, allowing many
more particles (atoms) to be considered
during
simulations.
The types of biological
activity that have been
investigated using
molecular
modeling include protein
folding, enzyme catalysis,
protein stability,
conformational
changes associated with
bio-molecular function, and
molecular
recognition
of proteins, DNA, and membrane
complexes.
Softwares
for bio-models
SPiM
The
Stochastic Pi Machine (SPiM) is a
simulator for the stochastic
pi-calculus that
can
be used to simulate models of
Biological systems. The
machine has been
formally
specified, and the
specification has been proved
correct with respect
to
the
calculus.
17.22
Computational
Basics Underlying Synthetic
Strategies
ScatterWeb.NET
SDK
ScatterWeb.NET
SDK is a new approach to
working with wireless
sensor
networks.
It hides the complexity of embedded
programming and makes it
easy to
handle
objects representing wireless
sensors.
5.
DRUG DESIGN
Drug
design is presently the most
prominent and visible
application of information
processing
in chemistry. Clearly this is
due to the large scientific
and economic
investment
necessary for the
development of a new drug.
Efforts are made to
combine
all
possible means for
developing an understanding of the
relationships between
structure
and biological activity.
Consequently, this is an area
where quantum
mechanical
and molecular mechanics calculations
are effectively
employed.
CONCLUSIONS
The
collected examples explain
the use of computational methods in
synthesis of
organic
compounds and also
extendable to inorganic and
energetic materials.
Researchers
are focusing on creating the
computational tools that will
enable
biologists
and others working in the
life sciences to better
understand and
predict
complex
processes in biological systems,
which could revolutionize
our
understanding
of disease, and lead to new
and faster insights into
entirely novel
therapies
and better vaccines.
REFERENCES
1.
E. J. Corey, Pure
Appl. Chern., 14
(1967) 19.
2.
S.1. Salley, J.
Am. Chem. Soc., 89
(1967) 6762.
3.
H. S. Rzepa, "Chemistry and the
World-Wide-Web", chapter in "The
Internet:
A
Guide for Chemists", Ed. S.
Bachrach, American Chemical
Society, 1995.
See
http:// www.ch.ic.ac.uk/
java/HyperSpec
4.
E. J. Corey and W. T. Wipke, Science,
166
(1969) 178.
5.
M. Bersohn and A. Esack, Chern.
Rev., 76
(1976) 269.
6.
E. J. Corey, R. D. Cramer, III, and W. J.
Howe, J.
Am. Chem. Soc., 94
(1972)
440
7.
S. Hanessian, J. Franco, G. Gagnon, D.
Laramee, and B.
Laruche,
J.
Chem. Inf. Cornput. Sci., 30 (1990)
413.
8.
I. Ugi and P. Gillespie, Angew.
Chem., Int. Ed. Engl., 10
(1971) 914.
9.
J. B. Hendrickson, J.
Am. Chem. Soc.,
93 (1971) 6847.
10.
I.
Dugundji and I. Ugi, Top.
Curr. Chem., 39
(1973) 19.
Synthetic
Strategies in Chemistry
17.23
11.
J. B. Hendrickson, D. L. Grier, and A. G.
Toczko, J.
Am. Chem. Soc., 107
(1985)
5228.
12.
J. B. Hendrickson and T. L. Sander,
Chen.
Eur. J., 1
(1995) 449.
13.
E. Differding and L. Ghosez, Tetrahedron
Lett., 26
(1985) 1647.
14.
N. Allinger, Encyclopedia of
Computational Chemistry, 1-5
(1997) 1320.
15.
N. Allinger, Encyclopedia of
Computational Chemistry, 1-5
(1997) 1580.
16.
P. Politzer and J. Murray,
Theoretical and Computational
Chemistry:
Energetic
Materials, Part 1. Decomposition, Crystal
and Molecular
Properties,
2003,
Elsevier Pubs.
Abbreviations
NMR
Nuclear Magnetic
Resonance
GAMESS-
The General Atomic and
Molecular Electronic Structure
System
MOPAC-
Molecular Orbital
PACkage
SYNGEN-
SYNthesis GENerator
SOS-Simulated
Organic Synthesis
SAS-Simulated
Analytical Synthesis
Table of Contents:
- INTRODUCTION TO SYNTHETIC STRATEGIES IN CHEMISTRY:POROUS MATERIALS
- SYNTHETIC METHODS BASED ON ACTIVATING THE REACTANT:HALOGENATION OF BENZENE
- METHODS BASED ON ACTIVATING THE REACTING SUBSTANCE:Experimental method
- SYNTHESIS OF MATERIALS BASED ON SOLUBILITY PRINCIPLE
- SOL-GEL TECHNIQUES:DEFINITIONS, GENERAL MECHANISM, INORGANIC ROUTE
- TEMPLATE BASED SYNTHESISSynthesis, Mechanism and Pathway
- MICROEMULSION TECHNIQUES:Significance of Packing Parameter
- SYNTHESIS BY SOLID STATE DECOMPOSITION:DECOMPOSITION METHODS
- NEWER SYNTHETIC STRATERGIES FOR NANOMATERIALS:Nanostructured Materials
- THE ROLE OF SYNTHESIS IN MATERIALS TECHNOLOGY:The Holy Bible
- ELECTROCHEMICAL SYNTHESIS:FEATURES OF ELECTROCHEMICAL SYNTHESIS
- NEWER REACTIONS AND PROCEDURES: CATALYTIC AND NONCATALYTIC
- SYNTHETIC STRATEGIES - FROM LABORATORY TO INDUSTRY
- SYNTHESIS OF CHEMICALS FROM CARBON DIOXIDE:Carbon dioxide - Dry Ice
- CARBOHYDRATES TO CHEMICALS:MONOSACCHARIDES
- SOME CONCEPTUAL DEVELOPMENTS IN SYNTHESIS IN CHEMISTRY
- COMPUTATIONAL BASICS UNDERLYING SYNTHETIC STRATEGIES